The work group has a long time experience in quasi elastic neutron scattering (QENS) on complex molecular liquids ranging from simple molecules such as short n-alkanes, pentafluoroanisole, and pentafluorotoluene to more complex molecules like long chain n-alkanes, co-enzymes Q0 and Q10, and liquid polyoxyethylenes. In all these studies we focus on an understanding of the diffusion mechanism at molecular level. The aim is to disentangle from the many different relaxation modes in such liquids the relevant molecular motions responsible for the primary step of the long range translational self-diffusion. For this purpose it is extremely helpful to complement the experimental results by state-of-the-art MD simulations, which are also conducted in our group. Precise parametrization of the force fields are indispensable for reliable results of the MD simulations. For our studies this is ensured by a detailed validation of the MD results by corresponding QENS data within the full space and time range relevant for the observed processes. The time range accessible by QENS and a corresponding visualization of trajectories calculated from MD simulations are displayed in Fig. 1.

Collective Intermolecular Motions Dominate the Picosecond Dynamics of Short Polymer Chains
What are the dynamics of short polymer chains taking place at the onset of molecular self-diffusion? It is this fundamental question which is studied in our group mainly by means of quasielastic neutron scattering (QENS) experiments and molecular dynamics (MD) simulations.
It is known that the single molecules show a subdiffusive behavior prior to a crossover to the freely-diffusive regime. This anomalous feature is contrary to the predictions of the simple, well-known Rouse theory, which predicts a freely-diffusive behavior of the molecular dynamics at all times. Several models have been proposed, accounting for the subdiffusive scaling of the molecules’ displacement. However, a detailed validation of the methods is needed to verify the true mechanism evoking molecular self-diffusion.
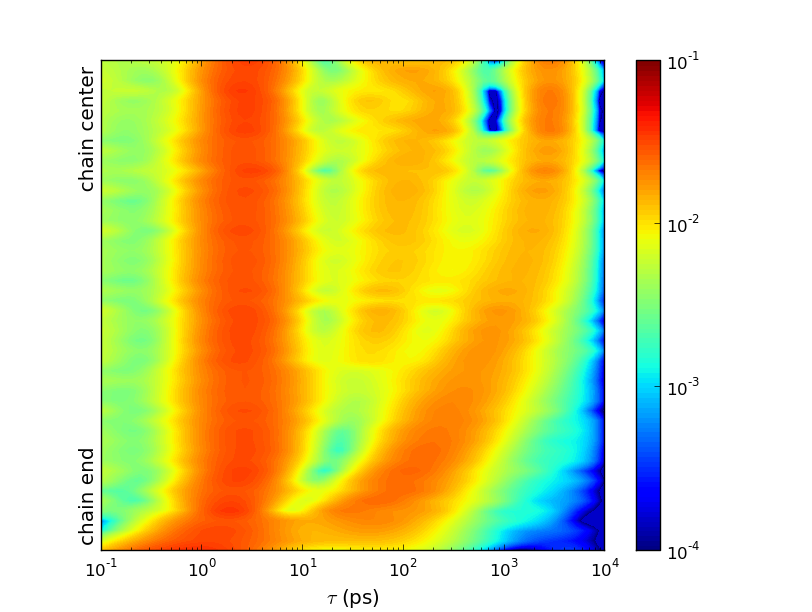
We recently presented the first experimentally validated observations, confirming that the atomic and molecular dynamics show viscoelastic properties, resulting in collective flow-like motions of large clusters of atoms. These flow patters strongly affect the dynamics of the single molecules.
Our results are based on extensive MD simulations of a C100H202 system, consisting of 4096 molecules (>1.2 million atoms). Complementary QENS measurements have been performed, and the resulting spectra were used to validate the MD simulations with high accuracy. Based on the outstanding agreement between the simulated and measured dynamics we used the simulations to characterize the prevailing molecular dynamics.
A detailed analysis of the chain relaxations revealed an unexpected mechanism, causing a molecule to change its conformation. By identifying the source of this relaxation a revolutionary picture of the molecular dynamics was found: Many neighboring atoms and molecules move collectively into similar directions, and it is the decay of these flow-like motions which causes the additional chain relaxation.
Based on these findings it is the goal of our group, to develop a fully consistent model of the transport behavior in molecular liquids. Accordingly, the characteristic motions in similar systems will be studied to gain a global picture of the underlying mechanism.